
Mark Zuckerberg, PDG de Meta, a annoncé ce 1er novembre que Meta AI avait créé un modèle qui prédit le repliement des protéines 60 fois plus rapidement que les modèles de pointe : ESM-2, qui compte 15 milliards de paramètres. La société l’a publié ainsi que « ESM (Evolutionary Scale Modelling) Metagenomic Atlas », une base de données de 617 millions de structures protéiques métagénomiques, alors que celle d‘AlphaFold 2, développé par DeepMind, qui prédit elle-aussi la structure tridimensionnelle d’une protéine à partir de sa séquence d’acides aminés, n’en compte « que » 200 millions.
Les protéines sont présentes dans toutes les cellules vivantes où elles assurent des fonctions essentielles. Les bâtonnets et les cônes dans nos yeux qui détectent la lumière et nous permettent de voir, les capteurs moléculaires qui sous-tendent l’ouïe et notre sens du toucher, les machines moléculaires complexes qui convertissent la lumière du soleil en énergie chimique dans les plantes, les enzymes qui décomposent le plastique, les anticorps qui nous protègent contre la maladie sont des exemples de protéines.
Les protéines métagénomiques qui se trouvent dans les microbes notamment dans le sol, l’air, au fond de l’océan et même à l’intérieur de notre intestin, sont beaucoup plus nombreuses que celles qui composent la vie animale et végétale, mais encore peu comprises.
La métagénomique utilise le séquençage génétique pour découvrir des protéines dans des échantillons provenant de ces environnements complexes. Elle a mis en évidence l’ampleur et la diversité incroyables de ces protéines, découvrant des milliards de séquences de protéines nouvelles, cataloguées dans de grandes bases de données compilées par des initiatives publiques telles que le NCBI, l’Institut européen de bioinformatique et le Joint Genome Institute, intégrant des études d’une communauté mondiale de chercheurs.
Selon Meta, « ESM Metagenomic Atlas, est le premier à couvrir les protéines métagénomiques de manière exhaustive et à grande échelle. Ces structures offrent une vision sans précédent de l’ampleur et de la diversité de la nature, et offrent le potentiel de nouvelles connaissances scientifiques et d’accélérer la découverte de protéines pour des applications pratiques dans des domaines tels que la médecine, la chimie verte, les applications environnementales et les énergies renouvelables. »
La création de cette « première vue d’ensemble de la “matière noire” de l’univers des protéines » a été rendue possible grâce au développement d’ESM-2, un modèle pour le repliement des protéines de Meta AI.
ESM-2, un modèle de langage de protéines de 15 milliards de paramètres
En 2019, Meta avait publié une étude démontrant que les modèles de langage apprennent les propriétés des protéines, telles que leur structure et leur fonction. En utilisant une forme d’apprentissage auto-supervisé connue sous le nom de modélisation du langage masqué, les chercheurs avaient formé un modèle de langage sur les séquences de millions de protéines naturelles. Avec cette approche, le modèle doit remplir correctement les blancs dans un passage de texte, tel que ” To __ or not to __, that is the ____ “. Ils ont formé un modèle de langage pour remplir les blancs dans une séquence de protéines, comme « GL_KKE_AHY_G » à travers des millions de protéines diverses.
Ils ont l’année suivante développé, ESM-1b, un modèle comptant environ 650 millions de paramètres qui est utilisé pour une variété d’applications, y compris pour aider les scientifiques à prédire l’évolution de la COVID-19 et à découvrir les causes génétiques de la maladie.
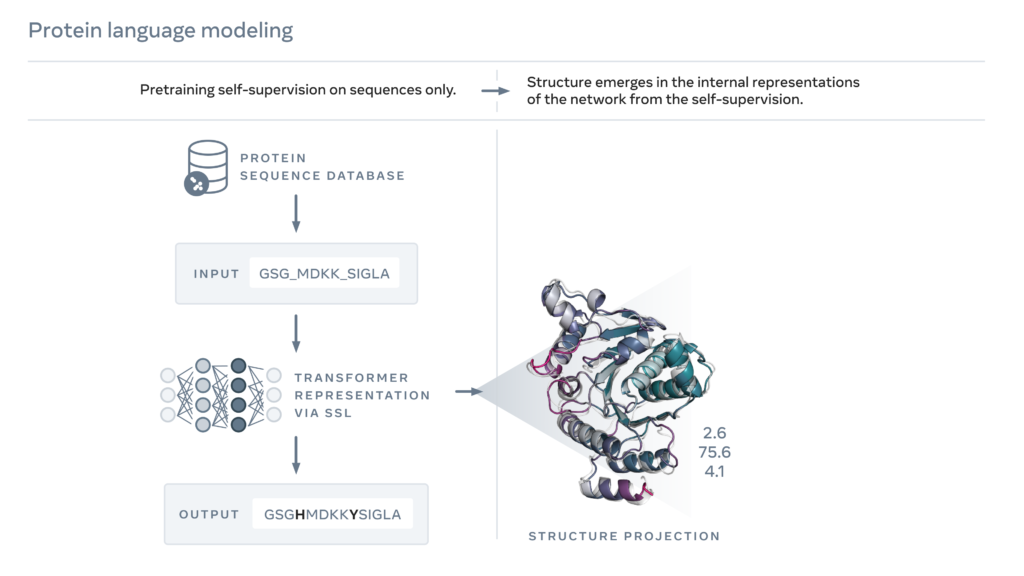
Les chercheurs ont étendu cette approche pour créer un modèle de langage protéique de nouvelle génération, ESM-2, qui, avec 15 milliards de paramètres, est le plus grand modèle de langage de protéines à ce jour. Ils ont constaté qu’à partir de 8 millions de paramètres, des informations émergeaient dans les représentations internes qui permettent de prédire la structure 3D à une résolution atomique.
Le réseau neuronal ESM-2 a permis de créer l’Atlas métagénomique ESM en prédisant 617 millions de structures à partir de la base de données de protéines MGnify90 en seulement deux semaines d’exploitation sur un cluster de 2 000 GPU. Tous deux devraient accélérer la découverte de nouveaux médicaments, aider à lutter contre les maladies et à développer une énergie propre.